1. 用途
分析Amber分子动力学轨迹,内容包括:RMSD、RMSF、B因子、氢键、回转半径、RDF以及距离、角度、二面角等几何指标的测量。
2. 预备知识
2.1 均方根偏差(Root Mean Square Deviation,RMSD)
分子模拟中采用RMSD来衡量构象差异程度或轨迹稳定程度。RMSD的定义如下:
其中,N是原子数,是原子的质量,是目标原子的坐标矢量,是参照原子的坐标矢量,是总质量。当RMSD不采用质量加权时,全部且。在Amber,RMSD的单位是埃米(Angstrom,Å)。
RMSD = 0.0意味着完美重叠,RMSD越大意味着目标分子偏离参照分子的程度越大。
计算RMSD需要指定参照分子,参照分子与目标分子在原子数和原子顺序上必须匹配。
在分子动力学模拟中,常用采用RMSD来快速判断某段轨迹是否达到平衡,以便进行后续模拟或分析。
RMSD的高度反映了轨迹相对于参照分子(通常选取轨迹第一帧,即初始状态)的整体偏离程度;但这不是至关重要的因素,因为采用不同的参照帧,高度和形状都会有所改变。RMSD曲线的形态和振幅则是值得重视的。当曲线有向上倾斜的趋势时,这表明体系构象可能发生某种显著运动(比如,蛋白Loop区“开合”折叠,RNA二级结构发生重大改变);当曲线在某个高度附近平稳振动时,这表明体系达到平衡,振幅大小反映了其振动的剧烈程度。
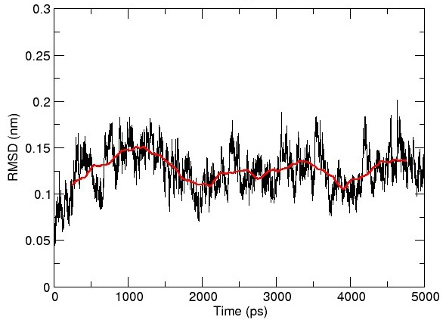
下图是典型的平稳的RMSD图(0.1 nm = 1 Å),曲线在1.3 Å附近波动,振幅保持在1.5 Å内。
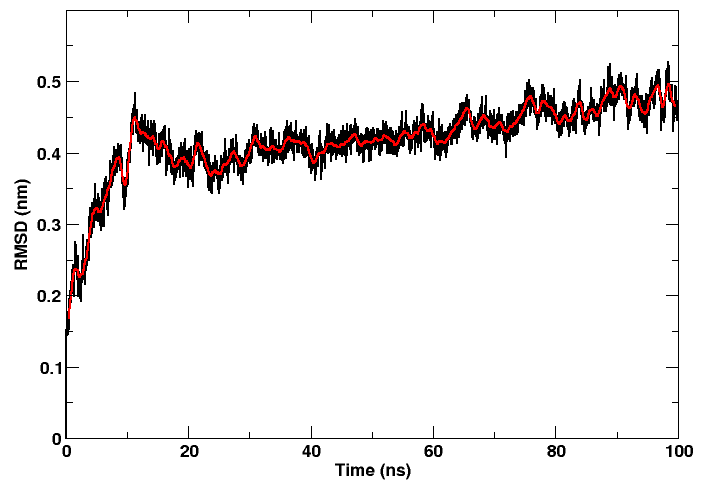
下图是蛋白骨架的RMSD曲线,从30 ns开始有缓慢抬升的趋势。模拟时间足够长(100 ns),它可能提示存在某种本质的蛋白骨架运动。
2.2 均方根涨落(Root Mean Square Fluctuation,RMSF)
RMSF计算每个原子相对于其平均位置的涨落(变化幅度),是衡量原子运动自由程度的指标,表征分子结构的柔性。其定义为:
其中,为时间,为时刻原子的位置,为该原子的平均位置,就是时刻原子相对于其平均位置的偏移量。在Amber中,RMSF的单位是Å。
当计算的是残基或一组原子的RMSF时,计算公式为:
其中,为原子的RMSF值,为其质量,即将各原子RMSF按质量进行加权平均。
RMSD与RMSF的区别:
- RMSD反映分子结构随时间的位置变化程度(偏移),横坐标是时间,纵坐标是整体偏移值;
- RMSF反映分子中各原子运动的自由程度(涨落),横坐标是残基(或原子),纵坐标是各自的涨落值。
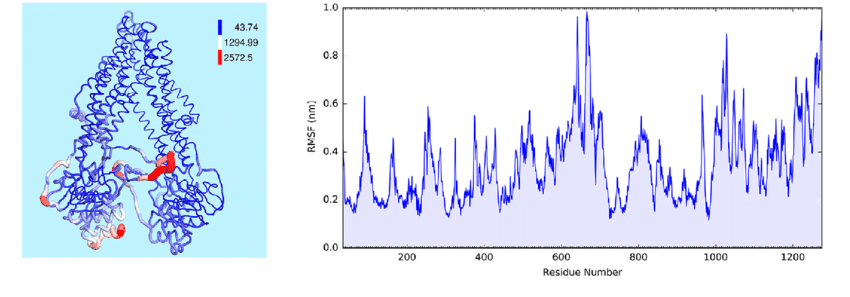
下方是典型的RMSF图(右图),将其显示在蛋白原子上(左图),可观察到特定区域有较高柔性,这些区域对蛋白发挥功能有重要作用:
2.3 B因子(B-factor)
温度因子(也称温度值,B因子,B值或Debye-Waller因子)是衡量原子位置不确定性的一种方法,体现了晶体中原子电子密度的“模糊度”(diffusion)。温度因子越高,相应部位的构象就越不稳定。B因子与RMSF的关系是:
在Amber中,B因子的单位为 Å2。
通过比较计算的B因子和晶体结构数据,可以考察模拟结果是否与晶体结构符合。
2.4 回转半径(Radius of Gyration)
回转半径(Radius of Gyration)描述体系原子沿着特定轴向的分布特征,可用于表征分子的紧密程度(compactness)。例如,可用于区分折叠的-螺旋结构和舒展的结构,或者考察体系的特征运动。其计算公式如下:
其中,是总质量,是原子的质量,是原子到全部原子的中心(通常为质心)的距离。
下图展示了原本体和突变体的回旋半径曲线,两者在高度和振幅方面都有显著差别,这表明突变后体系变得更“松散”且不稳定了。

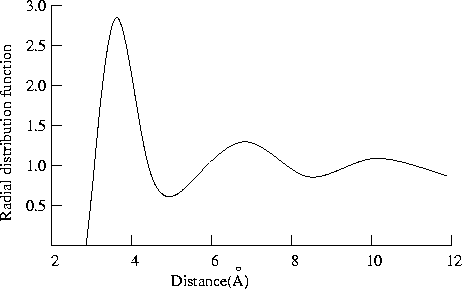
2.5 径向分布函数(Radial Distribution Function,RDF)
径向分布函数(Radial Distribution Function,RDF,也称对关联函数)是用来考察特定粒子周围其他粒子分布特征的一种统计方法。其定义是:在一个中心分子周围距离为处,分子的局部密度相对于本体密度的比值。公式为:
下面是典型的RDF曲线图:当距离较小时,RDF一直为0,随着距离增大,在某个临界点开始出现其他粒子,随后出现一个高峰,峰值远大于1,然后出现小于1的低谷,之后可能还会出现若干峰谷,在大距离区间,RDF趋近于1.0。
3. 入口
平台地址:(https://cloud.yinfotek.com/)
平台左侧菜单栏【计算方案】->【大方案】->【分子动力学】->【分析Amber轨迹】
4. 平台操作步骤
4.1 选择结构文件和设定采样范围
从平台云盘中选择拓扑文件和轨迹文件,点击【计算帧数】,获得各轨迹文件的帧数。建议选取经过【处理Amber轨迹】方案处理过的文件。
不同分析内容可能需要采用不同方式处理过的文件;下面会在具体的分析内容中详细介绍,若非特别注明,本教程采用来自【处理Amber轨迹】默认参数处理后的文件。
4.2 设置分析内容
按需要设置分析内容。
-
RMSD
为需要计算RMSD的结构填写Amber mask(详见《【分子动力学模拟(Amber 20)】》的预备知识),可填写多项,每项对应一条曲线。参照分子可选择轨迹的第N帧或上传文件。
例如,计算某蛋白1-266号残基的蛋白骨架(CA、C和N原子)RMSD,可在mask填写
:1-266@CA,C,N。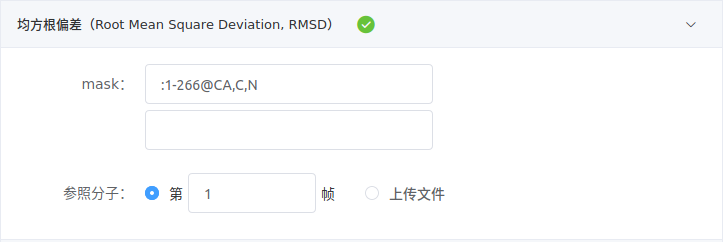
-
RMSF
为需要计算RMSF的结构填写Amber mask(详见《【分子动力学模拟(Amber 20)】》的预备知识)。 计算方式有三种:
-
按残基
得到每个残基的RMSF值,常用于评价生物大分子残基柔性。
-
按原子
得到每个原子的RMSF值。
-
按原子群
得到mask结构整体的RMSF值。
-
例如,计算蛋白1-266号残基的RMSF,可在mask填写
:1-266,选择按残基方式。

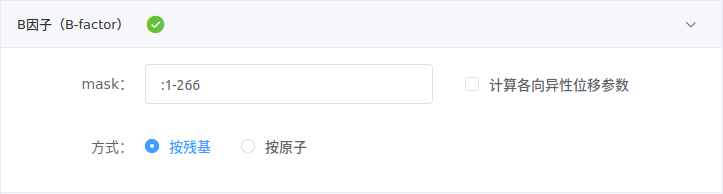
- B因子
B-factor的设置方式与RMSF类似,勾选计算各向异性位移参数可得到类似于PDB文件中ANISOU的文件。计算方式只提供两种:按残基和按原子,含义与RMSF类同。
例如,计算蛋白1-266号残基的B因子,可在mask填写
:1-266,选择按残基方式。
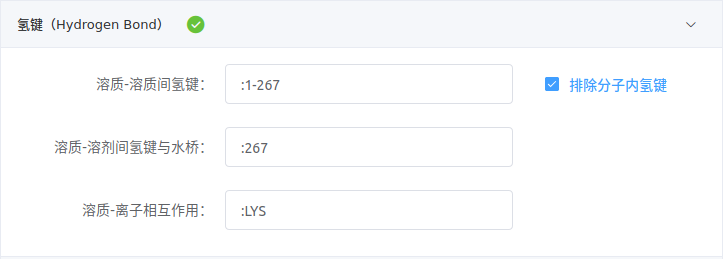
- 氢键
溶质-溶质间氢键用于分析溶质与溶质之间的氢键,需要填写氢键供体和受体的mask。分两种情况:
- 分子间氢键
分析独立分子之间的氢键,例如,受体蛋白与配体间的氢键、蛋白两链间的氢键。此时,应勾选排除分子内氢键以免分析蛋白链内残基间的氢键,产生过多不必要的信息。
例一、计算蛋白(1-266号残基)与配体(267号残基)之间的氢键,可填写
:1-267,并勾选排除分子内氢键。例二、计算配体(残基名为BAX)与周围7 Å以内的残基之间的氢键,可填写
:BAX<:7,并勾选排除分子内氢键。
- 分子内氢键
分析分子内的氢键,例如,蛋白链内某些残基之间的氢键。此时,应不要勾选排除分子内氢键。
注意,每条不相连的链(包括由序列间隙分隔成多段的链)均视为独立的分子。
例如,计算蛋白链(1-134号残基)内氢键,可填写
:1-134,不要勾选排除分子内氢键。
溶质-溶剂间氢键与水桥用于分析溶质和溶剂(水)之间的氢键和水桥,溶质-离子相互作用用于分析溶质和离子之间的相互作用。两项均只需填写溶质的mask。
注意,【处理Amber轨迹】方案的默认操作是删除水和离子。因此,进行该分析时,请确保轨迹文件中包含溶剂和离子。必要时,请提交新的任务进行单独分析。
例一、分析配体分子(267号残基)与溶剂水之间的氢键和水桥,可填写
:267。例二、分析所有赖氨酸残基(LYS)与溶剂离子(K+、Na+、Mg2+、Ca2+和Cl-1)之间的相互作用,可填写
:LYS。
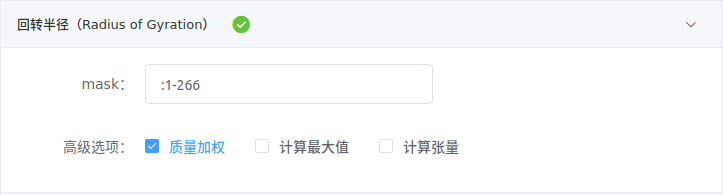
- 回转半径
填写需要计算回转半径的mask。默认对原子进行质量加权,勾选计算最大值将使用最大回转半径(而非均方根值),计算张量将计算回转半径张量。
例如,计算蛋白(1-266号残基)的回转半径,可在mask填写
:1-266。
- RDF
在溶剂mask填写需要计算RDF的原子的mask,此为必填项。若同时填写溶质mask,则以溶质每个原子为中心,计算将计算溶剂原子的RDF。
溶剂mask不一定是溶剂,也可以是任意原子,该命名只是方便区分。- 【处理Amber轨迹】方案的默认操作是删除水和离子。因此,若该分析涉及水或离子,请确保轨迹文件中包含这些分子。必要时,请单独提交任务进行本项分析。
- 顾名思义,RDF计算的是每对粒子的距离,计算量会随着粒子数量剧增,请注意设定适当的mask和轨迹采样范围。必要时,请单独提交任务进行本项分析。
这里提供了4种计算方式:
-
按原子:以
溶剂或溶质mask中的原子作为计算单元。 -
按原子群几何中心:以
溶剂或溶质mask中的原子群的几何中心作为计算单元。 -
按残基质心:以
溶剂或溶质mask中的每个残基的质心作为计算单元。 -
按分子质心:以
溶剂或溶质mask所指的每个分子的质心作为计算单元。
此外,需要确定距离区间宽度(图5的)和区间范围(图6的横坐标最大值)。
另外还有两个高级选项:
- 勾选
根据体积确定密度将根据轨迹中的体积自动计算密度,否则将采用固定的密度值(0.033456个分子/ Å3,对应于1.0 g/mL水的密度); - 勾选
排除分子内距离将忽略分子内距离,只计算分子间距离。
例如,计算配体(残基名为BAX)周围水分子的RDF,可在
溶剂mask和溶质mask分别填写:WAT@O和:BAX,对于溶剂,选择按原子方式计算,对于溶质,选择按残基质心,不勾选根据体积确定密度。

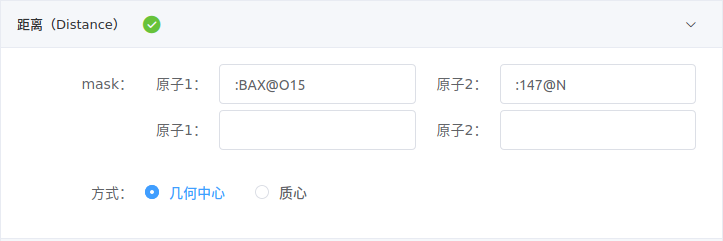
- 距离
在原子1和原子2填写要计算距离的mask,每组mask定义一个距离。若mask包含多个原子,将以原子群的几何中心或质心进行计算。
例如,计算配体(残基名为BAX)的O15原子和147号残基N原子之间的距离,可在
原子1和原子2分别填写:BAX@o15和:147@N,选择几何中心。
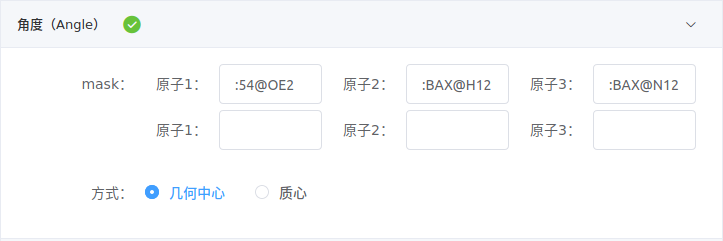
- 角度
与测量距离同理,在原子1、原子2和原子3中填写要计算角度的mask,每组mask定义一个角度。几何中心和质心的含义与上同。
- 二面角
与测量距离同理,在原子1、原子2、原子3和原子4中填写要计算二面角的mask,每组mask定义一个二面角。几何中心和质心的含义与上同。

4.3 点击【提交】,提交任务。
4.4 任务完成后,进入分析页面,可查看分析内容。内容按字面意思很好理解,因此不再赘述。
RMSD、RMSF、氢键、回转半径、距离、角度、二面角等随时间变化的统计指标,平台给出的横坐标为
Frame,但发表文章时通常写作Time (ps)或Time (ns)。换算关系取决于所选轨迹的采样间隔和每一帧代表的时间间隔。例如,原始轨迹1帧对应1 ps,如果采样间隔为10帧,则数据结果的1帧对应10 ps。氢键数据的详细解释见《【分子动力学模拟(Amber 20)】》文章的结果分析。
