1. 用途
检查并修复生物大分子PDB的结构问题,为分子对接、分子动力学模拟等计算提供良好的三维结构。
2. 预备知识
2. 1 PDB常见问题
大多数PDB文件或多或少存在着各种问题,在进行分子模拟时,首先需要修复这些问题,才能确保计算的可靠性。常见的问题有:
- 插入残基、重复残基、重复原子;
- 修饰氨基酸成为非标准氨基酸;
- 缺失残基(如:序列缺口)、缺失原子。
还有一些不算问题,但在分子动力学模拟中往往需要处理的方面:二硫键、酸碱氨基酸。
此外,X衍射晶体结构通常不含氢原子,但分子模拟往往需要补全。
2.2 插入残基
在研究不同物种来源的某种蛋白时,人们发现它们的序列之间存在一定联系(比如,序列进化)或存在某种重要的模式(比如,某些蛋白家族中的motif)。为了更好的体现这种联系、方便讨论和比较不同物种的结构或者为了符合某种外部标准,人们希望保留特定的残基编号而不严格递增编号。例如,在PDB编号为1IGY的晶体结构中,B链残基52和53之间插入了一个“52A”残基(A为插入码)。
分子动力学模拟通常要求所有残基编号唯一,且从1开始重新编号。
2.3 替代位置
通常情况下,PDB文件中每个残基的原子位置是唯一的,但在某些情况下,可能见到同一个原子有多个位置。大分子晶体结构是由许多包装到一个对称排列中的独立分子构成的。在有的晶体结构中,不同分子之间的构象存在些许差异。例如,位于蛋白表面的侧链可能在不同构象中来回折叠,底物结合在活性位点中的构象并非唯一,金属离子仅存在于个别分子中。在PDB文件中,这种情况以“替代位置”(alternate location)的方式记录下来(在残基名钱),各自出现的概率用占有率(occupancy)描述。
分子模拟要求清除多余的构象,仅保留一个确定的位置。通常,我们会选择occupancy最大的那个。
2.3 蛋白封端
当PDB文件中缺失残基时,会出现序列缺口(gap)。对分子动力学模拟而言,不做处理很可能会导致严重的问题。一种做法是将氨基和羧基分别处理成带电离子(NH3+和COO-),这对远离结合口袋的缺口是可以接受的。但更好的办法是添加帽子(cap)进行封端。常用的帽子是ACE和NME,分别加在N端和C端。注意:加帽封端因引起此后残基编号的改变。
2.4 质子化与去质子化
在特定pH下,碱性氨基酸会发生质子化,酸性氨基酸则发生去质子化,有时候这种质子状态对研究对象而言非常重要。在分子动力学模拟中,是通过不同残基名代表的结构状态来精细描述的。
-
组氨酸(HIS)
组氨酸有三种质子化状态:当氢原子在位氮原子时为HID,在位氮原子时为HIE,两个位置均带氢原子时为HIP。
一般来说,组氨酸侧链的pKa在6.0-6.5左右,在生理条件下(血浆=7.2-7.4)呈现电中性,为HID或HIE型,而在酸性环境下带正电,呈现HIP型。当然,在蛋白质内存在pKa位移效应,使其pKa发生偏移。因此,做分子模拟(尤其是分子动力学)之前最好做pKa分析以判定各个残基(尤其是组氨酸)的质子化状态。

-
天冬氨酸(ASP)
天冬氨酸为酸性氨基酸,通常以去质子化状态(带负电)存在,记为ASP;少数情况下呈中性,记为ASH。
-
谷氨酸(GLU)
谷氨酸也是酸性氨基酸,通常带负电,记为GLU;少数情况下呈中性,记为GLH。
-
赖氨酸(LYS)
赖氨酸是碱性氨基酸,通常以质子化状态(带正电)存在,记为LYS;少数情况下呈中性,记为LYN。
3. 入口
平台左侧菜单栏【计算方案】->【小工具】->【分子动力学】->【处理PDB结构(进阶版)】
4. 步骤
4.1 检查PDB
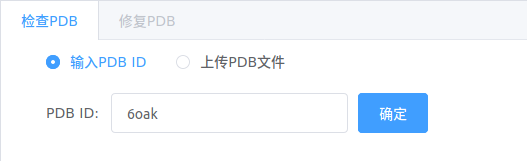
-
输入PDB ID或上传PDB文件,点击【确定】;
-
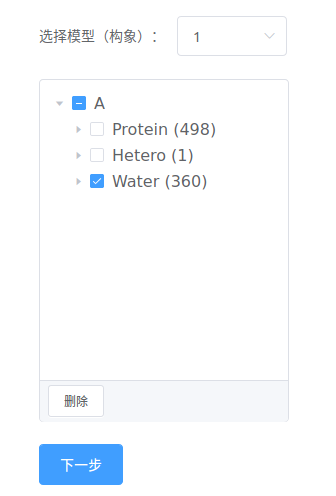
选择模型(构象)
通常,X衍射晶体结构文件中只有一个模型(构象),而NMR结构会有多个模型(构象)。
-
勾选需要删除的链或残基,点击【删除】,仅保留需要的结构;
勾选后,分子视图中会同步高亮显示。
-
点击【下一步】,稍等片刻,即返回结构检查信息。
4.2 修复PDB
-
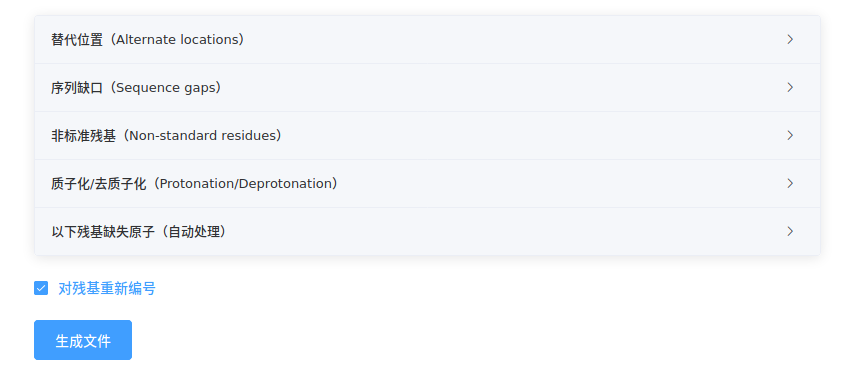
认真查看折叠卡片中陈列的结构问题(详见附录),根据实际需要进行处理;
目前,本工具检查7类问题,本页面仅显示当前PDB文件存在的问题。
-
勾选或去选对残基重新编号,点击【生成文件】,下载文件;
对于分子对接,为方便分析,通常不勾选对残基重新编号,以保持残基编号;
对于分子动力学模拟,残基编号总是重新编号,且无链名。因此,建议勾选对残基重新编号,以获得新旧编号对照表renumber.csv,方便后续分析。
此外,无论勾选与否,链名都会保留。

4.3 附录
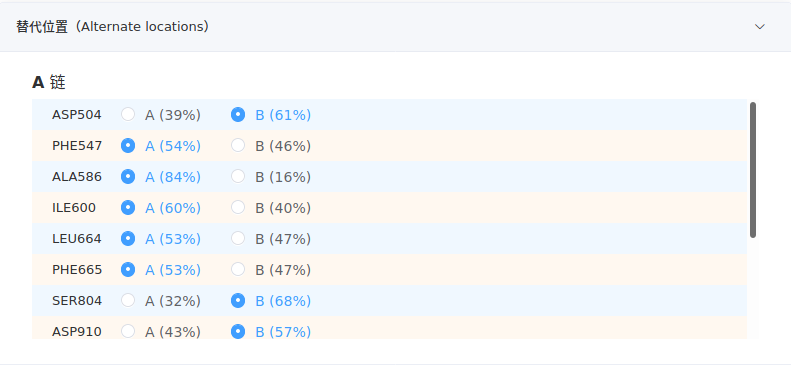
- 替代位置
该项列出存在不同构象的残基及各位置的占有率,默认选择占有率最大的,用户可以分析结构,选择最有利的构象。
若这些残基位于研究重点或附近的区域(例如,配体分子的结合口袋),有必要仔细观测各位置构象,选择合适的一个。
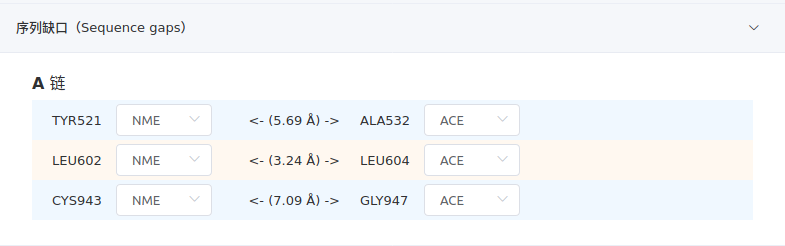
- 序列缺口
该项列出蛋白、核酸中缺失的残基所形成的序列缺口。对于蛋白质,默认采用NME和ACE封端,也可以选择NH3+和COO-;对于核酸,无封端帽子。每一组缺口的箭头中间是肽键长度。
应当注意,特殊情况下该缺口可能是虚报的。例如,当某氨基酸被修饰过,成为非标准氨基酸,因不被识别而产生缺口。因此,当存在“非标准残基”项时,应当检查是否存在(编号上)正好衔接缺口两端的残基。
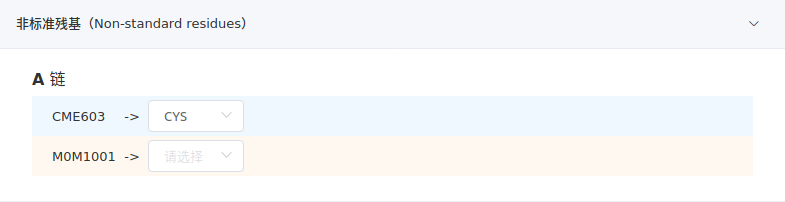
- 非标准残基
该项列出所有非标准氨基酸和碱基的残基,可能是修饰过的氨基酸或碱基,也可能是有机小分子,平台会尽力给出正确的猜测。如下图所示,CME603实际上是标准氨基酸CYS侧链S原子连接2-巯基乙醇产生的非标准氨基酸,箭头右侧自动选择CYS,表示要替换成CYS。而M0M1001是有机小分子,无对应的标准氨基酸,箭头右侧留空表示不做替换处理。
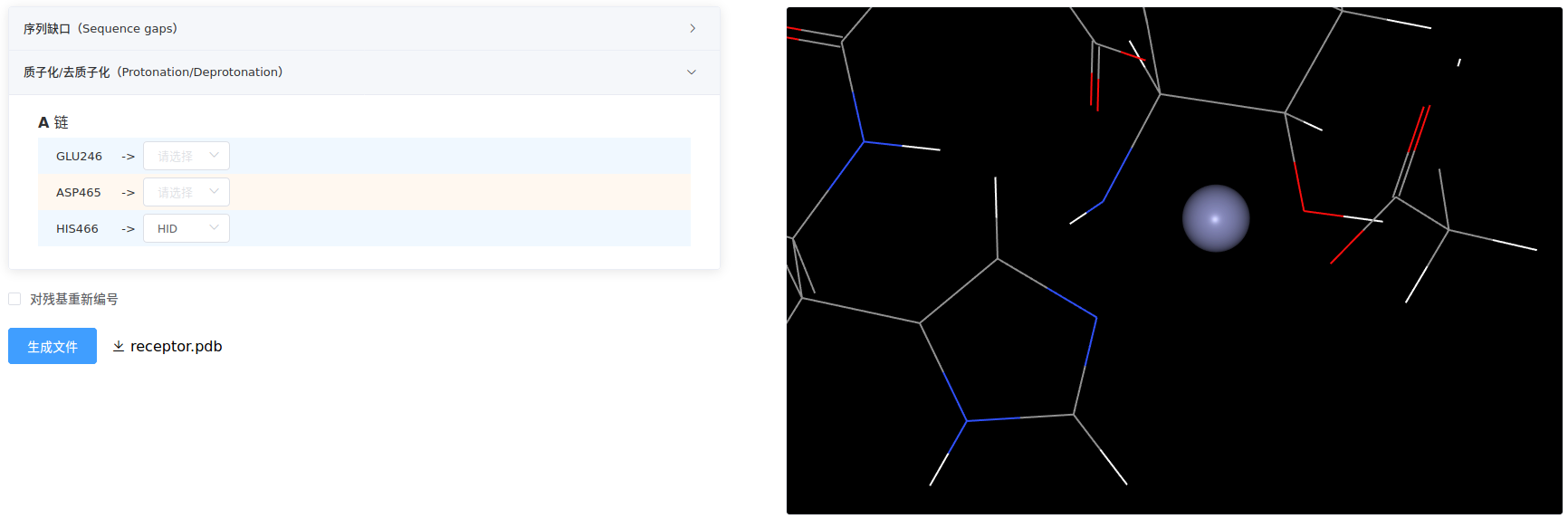
- 质子化/去质子化
该项列出所有酸碱氨基酸,用户可针对个别氨基酸进行精细调整。在某些情况下,尤其是活性口袋、催化位点、金属配位结构域等等重要区域,需要仔细分析调整。
例如,下图所示,不做选择时由程序自动质子化/去质子化,点击【生成文件】。观察发现Zn离子配位的HIS466在位连接有氢原子,与Zn发生原子碰撞,破坏了配位键。
需要将生物大分子的显示样式设为line,着色方案设为element。
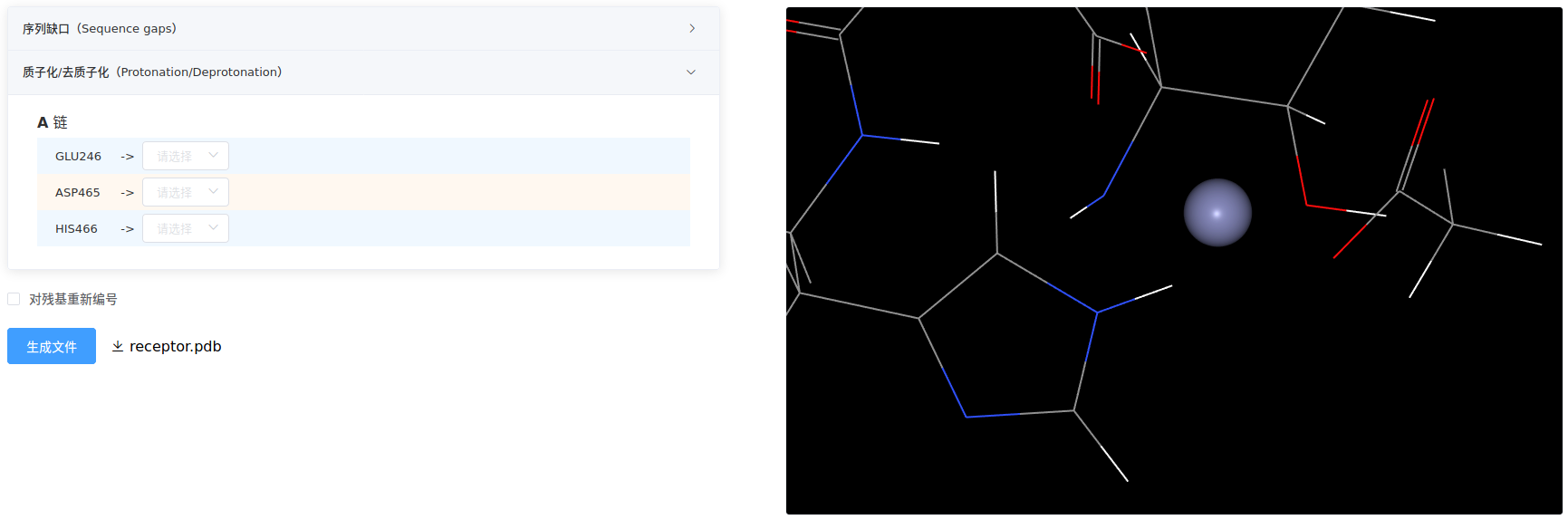
因此,需要把它调整为HID,再次点击【生成文件】,氢原子就连接在位,保证配位键。
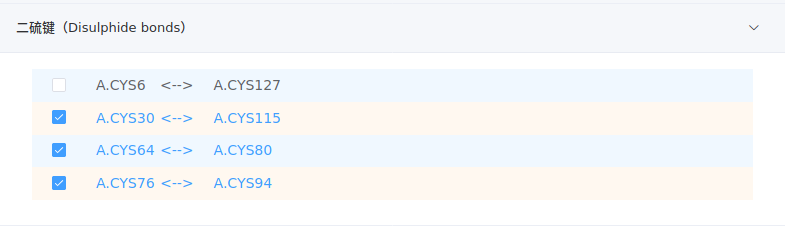
- 二硫键
该项列出所有被识别出的二硫键。对于分子动力学模拟,二硫键需要特殊的残基名(CYX)来标识;对于分子对接,可以不考虑二硫键,采用PDB的标准名称(CYS)即可。勾选的二硫键残基将被改名为CYX,没有勾选的则保持原名。
- 缺失原子的残基
该项列出不完整的氨基酸和碱基,并标记各自缺少的重原子(非氢原子)数目。无需用户操作,平台会自动处理。

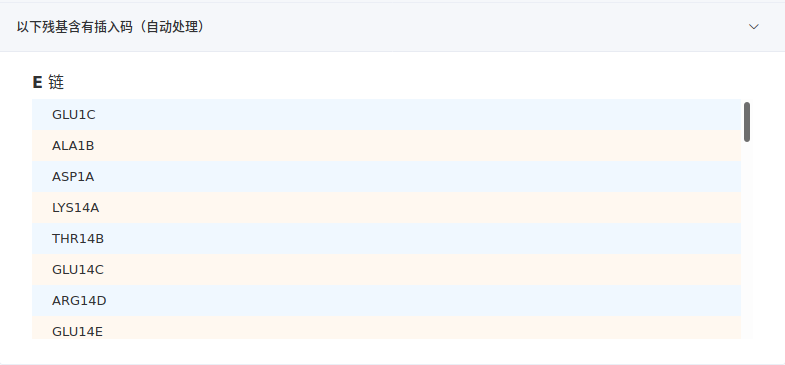
- 含有插入码的残基
该项列出含有插入码的残基。无需用户操作,平台会自动处理。
当勾选对残基重新编号,插入码会被删去,残基编号会从按顺序递增;当不勾选,插入码和残基编号都会保留。